病因
病因
病因:本病病因尚无定论。多数学者认为是常染色体隐性遗传性疾病。曾有一家9个同胞中5个患病和一家连续2代4例患病的报告。现代分子生物学技术也揭示Bartter综合征是由肾小管上皮细胞上的离子转运蛋白基因突变所引起。目前已发现婴儿型Batter综合征存在Na -K -2Cl-基因突变,该基因位于15q12-21,有16个外显子,编码1099个氨基酸,为Na -K -2Cl-通道,已发现20多种突变。经典型Bartter综合征系由CICNKB基因突变所致,该基因位于1q38,编码含687个氨基酸的细胞基底侧的Cl-通道,现已发现约20种突变类型。成人型Bartter综合征又称Batter-Gietlman综合征,系由噻嗪敏感的Na -K 通道基因(SCI12A3)突变所致,该基因定位于16q913,编码1021个氨基酸,已发现多达40种突变。此外还有一些病人中发现钾通道基因(ROWK)突变。因此Batter综合征可以认定为由上述几种离子通道基因突变引起的临床综合征。
发病机制
发病机制:本症的发病机制尚未完全阐明。有人就本综合征发病环节提出4种假说:
1.血管壁对ATI的反应有缺陷导致肾素生成增多和继发性醛固酮增多。
2.近端小管钠重吸收障碍导致钠负平衡;低钠饮食亦不能逆转肾性失钾。
3.前列腺素生成过多,使肾小管失钠,血钠减低从而激活肾素-血管紧张素系统。
4.髓襻升支厚壁段对氯化物转输障碍,使氯化物重吸收减少,钾排泄增多导致
低钾血症;
低钾血症刺激前列腺素E
2的生成,并使血浆肾素活性和血管紧张素Ⅰ升高。前列腺素E
2升高后血管对ATI不敏感,因而血压正常。
近年来的临床与实验研究对Bartter综合征发病机制的认识有了很大的进展,认为Bartter综合征是由于髓襻升支厚壁段穿上皮细胞Cl
-、Na
的转运障碍所致。目前对髓襻升支的几种离子通道蛋白的基因编码已经克隆出来,由于这些离子通道蛋
白发生了丧失功能的基因突变,致使离子转运功能发生障碍。正常肾单位髓襻升支厚壁段(图1)对Cl
-、Na
再吸收是由对布美他尼敏感的钠-钾-2氯运载体(bumetanide-sensitive sodium-potassium-2-chloride transporter,NKCC2)进行的。由于细胞内Na
与C1
-较细胞外低,NKCC
2将Na
、K
、2Cl
-运转入细胞内,仍维持电中性。上皮细胞的基侧膜上有Na
-K
-ATP酶能把过多的Na
泵出细胞外,进入血液。另外,还有肾脏特异性基侧氯通道(kidney specific base lateral channel,CIC-kb)把Cl
-泵出细胞外,经血液再吸收。髓襻升支厚壁段上的管腔膜上还有ATP调节钾通道(ATP-regulated potassium channel,ROMK)。NKCC2的转运速率是由ROMK对钾再循环进行调节,即ROMK为NKCC2提供有效的K
浓度,保证管腔的正电位。
基因研究推断,上述离子运载体蛋白或通道蛋白中任何一种发生突变,都可能出现离子转运障碍,从而导致Bartter综合征的发生。不同的通道蛋白或载体的缺陷可形成Bartter综合征的不同的亚型。目前认为,由于NKCC2功能丧失性突变,导致Na 、K 的再吸收障碍;ClCkb通道蛋白失活,限制了NKCC2运载体的转运速率,损害了K 的再循环过程对K 的再吸收。所以,只要上述环节中任何一种环节上发生了功能丧失性突变,都会削减上皮细胞电位差,减少上述离子重吸收的驱动力(图2)。
髓襻升支厚段再吸收Na
、Cl
-减少,细胞外液量轻度降低,继发高肾素、高醛固酮血症和肾小球旁器增生与肥大。由于氯化钠大量流经集合管,刺激泌H
、泌K
,加上高醛固酮血症,因而引起
低钾血症和
代谢性碱中毒。肾素-血管紧张素-醛固酮系统功能亢进,促进激肽、血管舒缓素生成,前列腺素生成增多,使血管对血管紧张素反应降低,血压保持正常,无水肿表现。最近研究发现,Bartter综合征患者单核细胞NO合成酶(ecNOS)mRNA水平呈高表达,尿中NO代谢产物NO
2-/NO
3-与cGMP平行升高,推测由于NO产生增多,减少血管张力,认为也是Bartter综合征患者血管对血管紧张素反应性降低的原因之一。有关ecNOS在Bartter综合征发病机制中的作用,尚需深入研究。
临床表现
临床表现:本病临床表现复杂多样,低钾症状为本征最重要和突出的表现,患者可突然或反复发作肌无力,肌无力也可为慢性持续性,但罕有肌
麻痹。其次为厌食、呕吐,
腹胀便秘,多尿烦渴。成人型最常见症状为肌无力(40%),其次为疲劳(21%)、抽搐(26%),较少见症状有轻瘫、感觉异常、
遗尿、夜间多尿、
便秘、恶心、呕吐,甚至
肠梗阻,有些患者还有嗜盐、醋或酸味腌菜,直立性
低血压,痛风以及高钙尿症,肾钙化,进行性肾功能衰竭,
维生素D缺乏病,镁缺乏,红细胞增多症等。少数严重病例可出现低钾所致的
心律失常,心电图显示
低钾血症表现,偶尔可引起肾盂和输尿管积水、巨结肠等所谓“空腔脏器扩大症”。甚者出现急性电解质紊乱,可表现为肠痉挛,低血钙可出现手足搐搦。儿童型最常见症状为生长延缓、发育障碍,身材矮小,
智力低下(占51%),其次为肌乏力(41%)、消瘦(3l%)、多尿(28%)、抽搐(26%)、烦渴(26%),并有特殊面貌异常如大头、突耳及下翻嘴等。至青春期可有突然生长加速使身材矮小明显减轻,机制未明。化验有低血钾、碱血症、低血钠、低血氯[婴幼儿可<(62±9)mmol/L]、尿钾高(>30mmol/L)。胎儿期Bartter综合征表现为间歇性发作的多尿,致孕22~24周出现
羊水过多,需反复抽羊水,以阻止
早产。得注意的是有少数病人无明显症状(10%小儿,37%成人),常因其他原因就诊时发现。
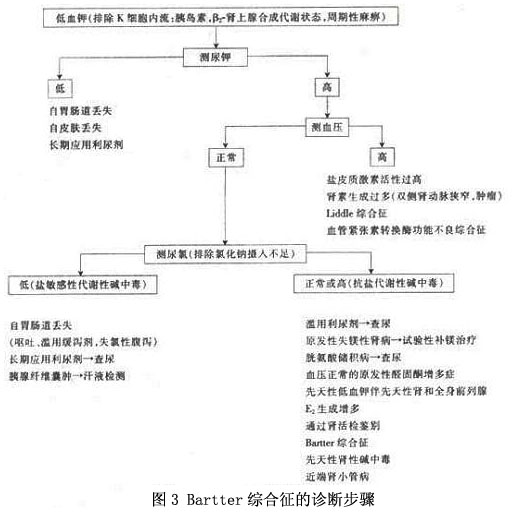
其他辅助检查
其他辅助检查:
1.KUB、IVP 显示肾盂及输尿管积水,肾钙化。
2.骨X线片 骨髓端闭合延迟。
3.胃肠X线检查 可见十二指肠扩张或其他空腔脏器扩张。
4.肾活检
(1)光镜下显示肾小球旁器(致密斑细胞)显著增生、肥大。
(2)电镜下肾小球旁器细胞内分泌颗粒增多。肾髓质内间质细胞增生、肥大;近端肾小管上皮细胞有空泡变性。病变进展时间长,除有肾小球旁细胞增生外,有肾动脉狭窄、管壁增厚,晚期肾小球出现玻璃样变,间质纤维化,肾小管萎缩。肾上腺球状带增生,皮质脂肪浸润。
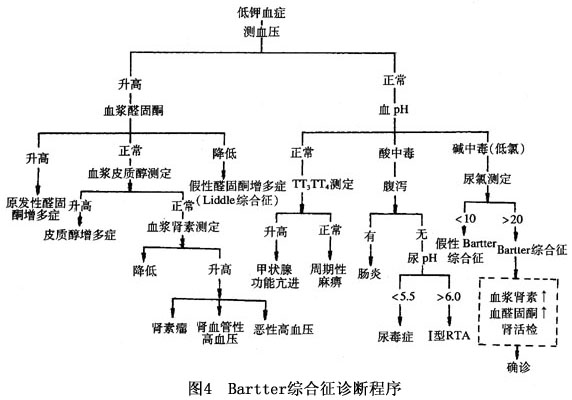
鉴别诊断
鉴别诊断:
1.本病应与假性Bartter综合征鉴别 所谓假性Bartter综合征为多种因素引起,常见的原因有利尿药的滥用、缓泻剂的应用、反复呕吐、长期低氯饮食、
肾性失镁、家族性氯化物性
腹泻。尿氯测定有助于鉴别,假性者尿氯多低于10mmol/L,真性者多大于10mmol/L。
2.其他 需与原发性醛固酮增多、
肾小管性酸中毒、Liddle综合征、Fanconi综合征伴
失盐性肾炎等鉴别。因各有其临床特点,鉴别不难(图4)。
例如:原发性醛固酮增多症有血压增高及血管紧张素Ⅱ降低;肾小管酸中毒为高氯性酸中毒,虽有
低钾血症而非碱中毒;原发性醛固酮增多症与Liddle综合征为先天性肾小管功能异常,无高肾素血症亦无高醛固酮血症,有
低钾血症和
代谢性碱中毒,而有高血压与钠潴留。Fanconi综合征伴
失盐性肾炎,以
低钠血症、高钠尿为主,可伴低血钾,根据各种疾病特有的临床及实验室特点均可仔细鉴别。
治疗
治疗:由于病因不明,目前尚无特殊治疗方法,主要以对症治疗为主。
1.饮食与活动 应食用含高钾的食物和饮料,如西红柿、香蕉、橙汁等。病人一般可正常参加基本活动,但是要避免活动过量引起脱水。以免脱水危险和低钾失衡导致功能性心脏功能紊乱。
2.药物治疗
(1)补充钾盐:适当控制钠入量并补充钾盐(儿童每天
氯化钾5~10mmol/kg)。10%
氯化钾溶液加入5%~10%
葡萄糖溶液500ml中缓慢静滴,同时口服保钾利尿药[螺内酯10~15mg/(kg・d),氨苯喋啶10mg/(kg・d),与它药合用时可适当减少剂量],用药过程中应注意监测血钾变化并及时调整钾盐用量。
(2)前列腺素合成抑制剂:常用药为吲哚美辛2mg/(kg・d),布洛芬3mg/(kg・d)或阿司匹林。
(3)应用抗肾素、血管紧张素类药物:如β-肾上腺素能阻滞药,普萘洛尔30~60mg/d,可降低肾素活性但不能纠
正肾性失钾。也可试用卡托普利(巯甲丙脯酸)(100mg/d)、普萘洛尔(心得安)等治疗。
(4)肾上腺切除术:文献报道4例曾行肾上腺切除术,3例有效,1例未见效果。